
第一原理計算ソフトウェア Advance/PHASE の基本機能
Advance/PHASE による第一原理計算から、ナノ材料(金属・半導体・絶縁体・磁性体・誘電体など)のさまざまな物性を解析・予測できます。
Advance/PHASE の特徴
- 平面波基底を用いた擬ポテンシャル法を採用
- 密度汎関数は、Local Density Approximation(LDA)および Generalized Gradient Approximation(GGA)、hybrid 汎関数が利用可能
- 相対論効果(スカラー近似)を考慮した擬ポテンシャルデータベースが付属:元素に応じ、Troullier-Martins 型のノルム保存形、Vanderbilt 型のウルトラソフト形、PAW を採用
- スピン分極効果を考慮することが可能
- バンド、k 点の 2 軸 MPI 並列化により、高い並列化効率を実現(Windows 版・Linux 版の両方で MPI 並列化に対応)
- 計算の準備と実行、および結果の解析を支援する GUI プログラムが付属
- 対応プラットフォームは Windows PC から地球シミュレータまで
- 開発元として責任を持ったユーザーサポート
- 卓越したコストパフォーマンス
Advance/PHASE の基本機能
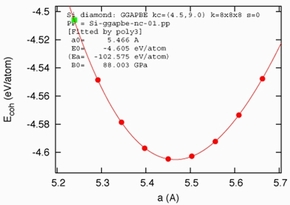
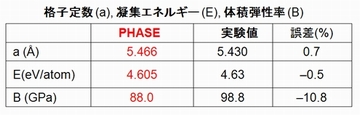
全エネルギー
系の全エネルギーを計算することができます。また、格子定数を変化させながら全エネルギーを計算することによって、絶対零度における格子定数や体積弾性率を求めることができます。
(現在のバージョンでは計算ユニットセルの自動最適化に対応しています。)
原子に作用する力
電子状態が得られると、原子に作用する力を求めることができます。力が作用する方向に原子を移動させることにより、(準)安定な原子配置が求まります。このような計算を構造最適化と呼びます。
下図に Pt(111)上の酸素原子を示します。初期状態として、酸素原子を適当な位置に配置します(右図)。構造最適化を行うと、酸素原子が安定な位置に移動し、吸着構造が求まります(左図)。
図中の矢印は、原子に作用する力を表しています。特定の原子のみを可動にすることができます。ここでは酸素原子の位置のみを最適化しました。

さらに、NVE および NVT 集合の分子動力学シミュレーションを行うことも可能です。
電子状態密度
シリコンの電子状態密度を示します。
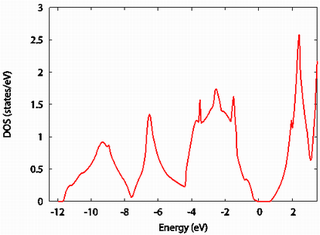
次に、原子欠損を含むシリコン結晶(シリコン原子 63 原子に対して空孔が一つ)の状態密度を示します。上図と非常に良く似ていますが、こちらにのみフェルミエネルギー付近に特徴的なピーク(青点線)が存在します。
このピークは、原子欠損に起因するものであろうと予想できます。より詳しい解析はこちらをご覧ください。なお、縦軸のスケールの違いは、計算している電子数の差によるものであり、本質的ではありません。
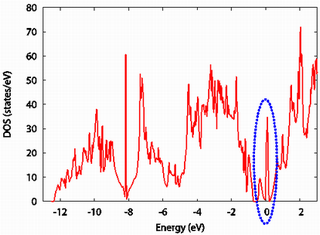
ここでは空孔を導入しましたが、不純物を導入するとそのドーピング効果を、状態密度から評価することができます。また、原子分割局所状態密度、層分割局所状態密度、射影状態密度も計算できます。
バンド構造
結晶のバンド構造を計算することができます。バンド構造図の横軸を意味する対称線(k点)は、GUI で指定できます(左図)。 右図はカリウムのバンド構造です。
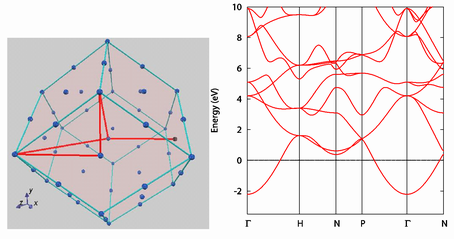
電荷密度分布
電荷(価電子)密度分布を可視化します。全価電子密度だけでなく、(非占有状態も含めた)任意のエネルギー準位に対応する電荷密度(部分電荷密度)を可視化することもできます。
グラファイトの電荷密度を下図に示します。左は全価電子密度です。原子間の結合に沿って、電荷密度の高い領域が存在しています。一方、右はフェルミエネルギー近傍の部分電荷密度です。π 電子的な電子状態が確認できます。
さらに本機能を応用することにより、STM像のシミュレーションが可能です。
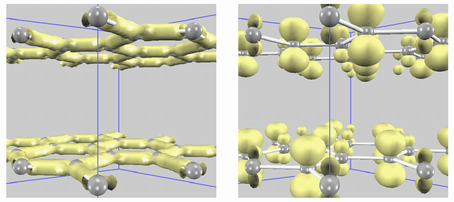
スピン分極効果
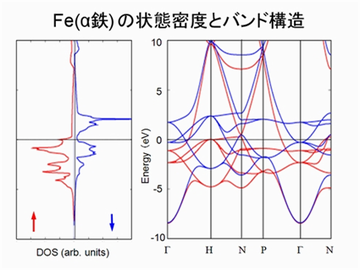
スピン分極を考慮した計算ができます。結晶構造と磁気秩序が異なる鉄(α、γ、δ)のエネルギーを、その体積を変化させながら計算しました。その結果から、α 鉄(体心立方格子の強磁性)が最も安定であることが分かります。
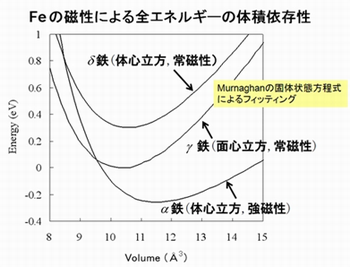
状態密度およびバンド構造は、アップスピンとダウンスピンを区別して計算されます。
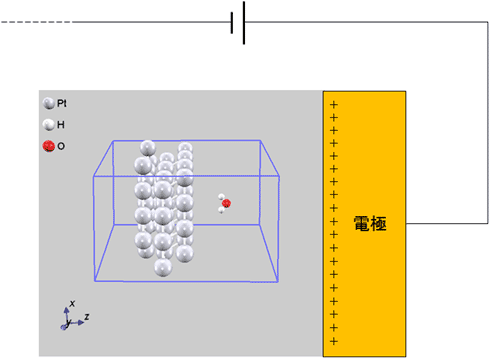
Effective Screening Medium(ESM)法
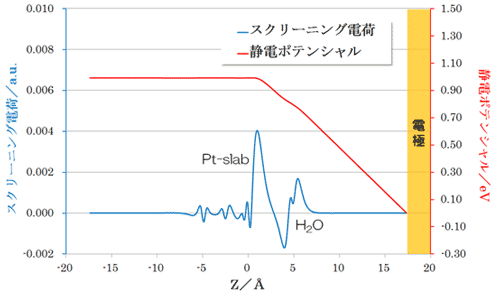
ESM 法により、電場を印加した計算が可能です。スクリーニング電荷をドープすることで、白金表面に静電場が発生することが確認されます。
より高度な解析機能、およびこれらの応用事例はこちらをご覧ください。